Глава II. Геохимические особенности элементов
Теперь можно считать уже твердо установленным, что сочетание химических элементов и их пространственное распределение при процессах минералообразования, происходящих в земной коре, находятся в причинной зависимости от свойств, обусловленных строением и размерами атомов и ионов самих элементов. Поэтому естественно, что внимание ученых давно уже началн привлекать вопросы классификации химических элементов, распространенных в земной коре, на основе таблицы периодической системы Менделеева. Из всех предложенных такого рода таблиц наиболее рациональна геохимическая таблица А. Н. Заварицкого, которая наиболее близко отражает естественные группировки химических элементов по их роли в земной коре, устанавливаемой путем изучения минерального состава природных геологических тел, их строения и возрастных взаимоотношений отдельных групп минералов.
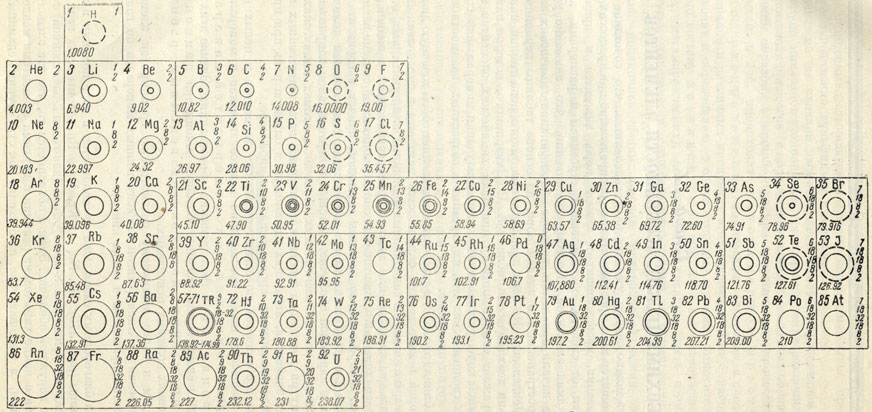
Рис. 370. Геохимическая таблица элементов. По А. Н. Заварицкому. Цифры перед названием элементов означают их порядковые номера; колонии цифр после знака элемента - числа электронов в каждой оболочке данного атома; цифры под кружками слева - атомные веса элементов. Кружки, показанные тонкими линиями, отвечают размерам атомов в данном масштабе; кружки, изображенные жирными сплошными линиями, соответствуют размерам катионов, а кружки, показанные пунктирными линиями, - размерам анионов
Эта таблица (рис. 370) в сущности представляет собой точечную диаграмму, в которой в виде тонких окружностей показаны в определенном масштабе размеры атомов, а в виде жирных (сплошных или пунктирных) окружностей - размеры катионов или анионов, распространенных в земной коре. Порядок расположения элементов отвечает периодической системе элементов Менделеева, построенной по длинным периодам, причем в горизонтальном направлении элементы сменяются в соответствии с изменением количества электронов в наружной оболочке атомов, а в вертикальном направлении - в соответствии с числом электронных оболочек в атомах химических элементов. Внутри диаграммы линиями оконтурены поля естественных групп химических элементов, играющих одинаковую и близкую роль при процессах минералообразования, протекающих в земной коре.
Знание особенностей поведения отдельных элементов и целых групп при этих процессах имеет чрезвычайно важное значение в минералогии, особенно для понимания закономерностей минеральных ассоциаций в геологических образованиях. Знание этих закономерностей, в свою очередь, оказывает большую помощь при рациональном направлении поисково-разведочных работ на месторождениях полезных ископаемых.
Перейдем к более детальному рассмотрению группировок элементов в геохимической таблице (список наиболее важных минералов по главнейшим элементам приведены в конце книги).
Группа благородных газов. Она располагается в виде колонки в левой части таблицы (рис. 370): Не, Ne, Аr Кг, Хе и Rn. Атомы всех этих элементов обладают устойчивой 8-электронной (для гелия 2-электронной) конфигурацией в наружной оболочке. Этим и объясняется то, что эти элементы не вступают ни в какие химические реакции с другими элементами менделеевской таблицы, а следовательно, и не входят как структурные единицы в состав кристаллических тел. В ряде минералов гелий устанавливается лишь в виде продукта радиоактивного распада в окклюдированном состоянии. В атмосфере и во многих газообразных выделениях из минеральных источников, а также в газовых струях из недр земной коры элементы благородных газов находятся в определенном соотношении (за исключением гелия и радона). Неравномерное содержание двух последних элементов в природных газах объясняют дополнительным образованием их путем распада радиоактивных элементов.
Группа главных элементов горных пород. Сюда относятся следующие элементы: Li, Be, Na, Mg, Al, Si, K, Ca, Rb, Sr, Cs и Ba. Все они образуют устойчивые ионы с 8-электронной наружной оболочкой (типа благородных газов) и отчасти ионы с двумя электронами во внешней оболочке (типа гелия). На рис. 370 видно, что эта группа элементов примыкает к предыдущей с правой стороны.
Главнейшее значение из них имеют Na, Mg, Al, Si, К и Са, входящие в состав многочисленных породообразующих минералов изверженных пород (нолевые шпаты, кварц, пироксены, амфиболы, слюды, нефелин, лейцит, оливин), метаморфических пород (хлориты, эпидот, скаполиты, серпентин, тальк, серицит и др.) и, наконец, осадочных пород (минералы глин, глинистых сланцев, мергелей, карбонатных толщ и др.). Насколько велика роль всех этих элементов в составе земной коры, можно судить по тому, что на долю главных породообразующих минералов приходится около 95% (повесу) всей массы земной коры. Следует добавить, что кварц, а также карбонаты Са и Mg являются главными нерудными минералами многочисленных жильных рудных месторождений.
Снизу к перечисленным элементам примыкают Rb, Sr, Cs и Ba-элементы с более низкими кларками и, как мы видели, входящие в виде изоморфных примесей главным образом в минералы группы полевых шпатов. Рубидий и барий часто устанавливаются в калиевых полевых шпатах (ортоклазе и микроклине). В случае значительных концентраций бария образуются гиалофаны (кали-бариевые полевые шпаты)*. Стронций очень часто обнаруживается в виде изоморфной примеси в плагиоклазах (особенно в нефелиновых сиенитах). Лишь цезий, ион которого обладает наиболее крупными размерами, склонен давать самостоятельные скопления в виде поллуцита в пегматитовых образованиях.
*(В гидротермальных рудных месторождениях барий распространен в виде безводного устойчивого сульфата-барита. )
Сверху в этой группе располагаются элементы Li и Be, обладающие малыми размерами ионов и потому также сосредоточивающиеся в остаточных магматических растворах, с помощью которых образуются пегматиты. Литий входит в состав таких минералов, как сподумен, литиевые слюды, литиевые фосфаты и др. Бериллий, как мы знаем, в наибольших количествах содержится в берилле, хризоберилле, фенаките и ряде второстепенных минералов, образующихся большей частью в тех же условиях.
Водород. В геохимической таблице элементов он занимает совершенно особое положение. В атомном состоянии в виде газа этот элемент образуется в сильно восстановительных условиях. Таковы находки его в ультраосновных изверженных породах - дунитах Нижне-Тагильского района на Урале, в месторождениях каменных углей, нефтей в смеси с другими газами (азотом, метаном), а также в некоторых соляных залежах. В окислительной обстановке атом водорода, теряя единственный электрон, превращается в "голое" ядро столь ничтожных размеров, что в кристаллических структурах минералов, как мы неоднократно убеждались, он не требует для себя какого-либо особого пространства, входя обычно в электронную сферу ионов кислорода. С его помощью образуется активный ион [ОН]1-, равновеликий с ионом F1- и потому заменяющий его во многих минералах. Как мы знаем, гидроксил входит в состав многих основных кислородных солей: силикатов (амфиболов, слюд и др.), фосфатов, сульфатов, карбонатов, а также гидроокислов (брусит, гидраргиллит и др.). Кислые соли в природе имеют очень ограниченное распространение.
При вхождении двух протонов Н1+ в ион кислорода образуется электрически нейтральная молекула Н2O. Она входит в состав водных твердых соединений, в которых удерживается слабыми связями, и является главной составной частью водных бассейнов, минеральных источников и вулканических эксгаляций.
Группа минерализаторов или элементов магматических эманации. Сюда принадлежат остальные элементы II и III периодов менделеевской таблицы: В, С, N, О, F, Р, S и Сl. Характернейшей особенностью этих элементов, известных под названием минерализаторов, является то, что все они образуют простые или комплексные анионы, играющие важнейшую роль в образовании самых различных солей при процессах минералообразования. Другая, не менее важная их особенность заключается в том, что большинство этих анионов с многочисленными катионами образует относительно легко растворимые и летучие (при уменьшении внешнего давления) соединения и, таким образом, играет огромную роль в переносе и последующей концентрации многих ценных металлов, характеризующих рудные месторождения как постмагматические образования. В силу этого магмы, первоначально содержащие летучие компоненты, при кристаллизации освобождаются от них. Поэтому изверженные горные породы, возникающие при затвердевании, как правило, очень бедны летучими соединениями.
Вместе с Н2O эти элементы, особенно В, F, Cl, S и С (в виде СO2), нередко в значительных количествах выделяются в виде газовой фазы вулканических эксгаляций. В глубинных условиях, вернее в условиях умеренных глубин, они часто оказывают сильное метаморфизующее воздействие на боковые породы, вмещающие массивы изверженных пород, с образованием грейзенов, скарнов и других контактово-метаморфических пород. На больших глубинах в среде магматических массивов они, очевидно, способствуют возникновению крупнокристаллических пегматитовых тел. В экзогенных процессах при их участии образуются многочисленные месторождения неметаллических полезных ископаемых (различных солей, боратов, фосфатов, самородной серы, каустобиолитов и др.).
Среди элементов рассматриваемой группы несколько особое положение занимают N и О, которые в атомарном состоянии входят главным образом в состав атмосферы (воздуха). Сравнительно небольшая часть азота устанавливается в газовых струях вулканических выделений, или в генетической связи с нефтяными месторождениями. Изредка образует нитраты на месторождениях селитры Этим подчеркивается в основном инертное поведение азота в земной коре.
Зато кислород играет исключительно активную роль в процессах минералообразованпя. Этот элемент в виде иона О2- и в меньшей степени в виде гидроксила [ОН]1- является главной составной частью окислов и кислородных солей (силикатов, фосфатов, сульфатов и т. д.), которые, как мы видели, имеют подавляющее значение в составе земной коры. Количество кислорода в ней по весу составляет 49,13%, а в объемном исчислении - 91,8% (крупные анионы О2- занимают господствующее положение по объему во всех кристаллических структурах кислородных соединений).
Очень важно подчеркнуть, что в литосфере по мере приближения к земной поверхности количество свободного кислорода в общем постепенно увеличивается. Особенно об этом можно судить по условиям нахождения тех минералов и минеральных ассоциаций, которые содержат металлы, способные давать несколько ионов разной валентности в зависимости от степени окислительного потенциала среды (например, в окислах FeO, Fe3O4 и Fe2O3 или МnО, Мn2O3, Мn3O4 и МnO2). При изучении состава горных пород, образовавшихся в различных условиях глубинности в земной коре, давно уже было доказано, например, что на большой глубине железо главным образом входит в состав силикатов в виде закиси или обособляется в виде магнетита. Гематит, как более богатый кислородом окисел железа, в этих условиях не образуется. Зато он гораздо более обычен в горных породах и рудных месторождениях, образовавшихся в условиях ближе к земной поверхности.
Кроме того, изучение парагенетических ассоциаций минералов в эндогенных горных породах и рудных месторождениях приводит к выводу о том, что по мере развития процесса минералообразования остаточные расплавы или растворы, как правило, несколько обогащаются кислородом, о чем можно судить по нередко устанавливаемым последовательным сменам парагенетических групп минералов, содержащих увеличивающееся количество кислорода. Например, в диференцированных дунито-пироксенито-габбровых массивах в главных породообразующих силикатах (соответственно в оливине, пироксенах, плагиоклазах) отношение числа ионов кислорода к числу катионов возрастает от оливина к плагиоклазам*. При этом некоторые элементы появляются в виде катионов более высших валентностей.
*(Оливиновые породы (дуниты) кристаллизуются раньше, нежели лироксениты и габбро (пироксено-плагиоклазовые породы). )
Нередко то же самое устанавливается в богатых кислородными соединениями рудных месторождениях. В последних стадиях минералообразования возникают более богатые кислородом минералы: гематит вместо магнетита и сидерита или вообще кислородные соединения вместо сульфидов тех же металлов и т. д. Известны также месторождения, в которых смены пара генетических ассоциаций минералов свидетельствуют о неоднократном колебании режима кислорода в ту и другую стороны в процессе рудообразования особенно при наложении новых стадий минерализации.
На самой поверхности на границе с атмосферой, богатой атомарным кислородом (23,1% по весу), происходит энергичное окисление эндогенных образований, главным образом сульфидов и им подобных соединений, а также кислородных соединений, содержащих Fe, Mn, V и др. в низших степенях валентности. При этом в качестве новообразований возникают окислы, гидроокислы, сульфаты, отчасти карбонаты и другие богатые кислородом соединения. Зона окисления, как известно, ограничивается снизу уровнем грунтовых вод. Ниже этого уровня хотя и устанавливаются признаки окисления, но они по степени проявления уже весьма значительно уступают предыдущим. В глубинных условиях в земной коре концентрация "свободного" (атомарного) кислорода в миллиарды раз ниже, чем в воздухе.
Сера является чрезвычайно интересным элементом в геохимическом отношении. Прежде всего следует подчеркнуть, что при участии серы осуществляется концентрация подавляющего большинства металлогенных элементов (см. правую часть таблицы, рис. 370) при образовании гидротермальных рудных месторождений. Почти все тяжелые металлы легко дают труднорастворимые соединения с ионами серы, тогда как сульфиды щелочей и щелочных земель, наоборот, легко растворимы в воде.
При этом, так же как и для окислов металлов, мы имеем для некоторых элементов сернистые соединения с различным отношением металл : сера, например, FeS (пирротин) и FeS2 (пирит, марказит), или Ni3S2 (хизлевудит), NiS (миллерит), Ni3S4 (полидимит) и NiS2 (ваэсит), или AsS (реальгар) и As2S3 (аурипигмент) и др. Эти различия в соотношениях металла и серы, несомненно, говорят о том, что их образование связано с различием концентрации серы в растворах в момент кристаллизации. Изучение парагенезиса минералов с учетом различия степени "сернистости" сульфидов металлов позволяет устанавливать определенные закономерности в последовательности образования минералов в сульфидных месторождениях, зависящие от режима серы и кислорода в растворах.
От кислорода сера, как элемент, отличается той геохимической особенностью, что в природных условиях она образует несколько ионов различной валентности, что имеет очень большое значение-в минералообразовании. В порядке возрастания степени окисления ионов; серы мы можем написать следующий ряд:
S2-→[S2]2-→So→S4+→S6+.
Образующийся в растворах анион S2-, как продукт электролитической диссоциации растворенного в них H2S устойчив, как можно видеть по его положению в этом ряду, в относительно более восстановительной обстановке реакций, в которой образуются сульфиды (FeS, Cu2S, SnS, PbS, ZnS и др.).
Спаренный анион [S2]2-, как мы знаем, входит в состав таких сульфидов, как пирит, марказит, ковеллин. Так как здесь отрицательный заряд (2-) относится к двум ионам серы, то, следовательно, на каждый ион серы приходится по одному отрицательному заряду, т. е. вдвое меньше, чем для предыдущего аниона S2-, что говорит об окислении иона *. Стало быть, пирит, содержащий в кристаллической структуре спаренные ионы [S2]2-, образуется в более окислительных условиях, чем пирротин.
*(Напомним, что потеря ионом или атомом электронов, сопровождающаяся уменьшением их размеров, свидетельствует об окислении (например, Feo→Fe2+→Fe3+ ). Наоборот, приобретение ионом электронов означает восстановление его (например, S6+→S4+→S0). )
Дальнейшее окисление ионов серы ведет к образованию электрически нейтрального атома SO, который при высоких температурах представляет собой газ, состоящий из молекул двухатомной серы, а при низких температурах (ниже 160°) молекула серы состоит уже из восьми атомов и кристаллизуется в виде самородной серы (при температуре ниже 120°).
Следующие ионы серы представлены уже катионами S4+ и S6+, возникающими в явно окислительной обстановке. Первый из них в природных условиях образует электрически нейтральную молекулу SO2 (сернистый газ, часто устанавливаемый в вулканических эксгаляциях), а второй - комплексный анион [SO4]2-,входящий в состав сульфатов (барита, целестина, ангидрита и др.). Поэтому не случайно, что сульфаты в глубинных условиях, как правило, не встречаются, за исключением тех случаев, когда их образование связано с остаточными обогащенными кислородом растворами. Зато они чрезвычайно широко распространены в экзогенных образованиях {в коре выветривания сульфидных месторождений, в осадочных гипсоносных толщах, в соляных озерах, богатых сульфатами щелочей и щелочных земель), в районах вулканической деятельности.
Галоиды фтор и хлор играют важную роль в переносе и концентрации ряда металлогенных элементов. О том, что эти элементы действительно присутствуют в числе легколетучих составных частей магм, говорит наличие их в весьма существенных количествах в большинстве эксгаляций, выделяющихся при вулканических извержениях. При этом, судя по фтор - и хлорсодержащим минеральным ассоциациям, которые возникают в постмагматических образованиях в различных по составу горных породах, содержание фтора как будто бы более свойственно кислым изверженным породам (грейзены и пегматиты с фторсодержащими минералами - топазом, слюдами, флюоритом и др.), а содержание хлора - основным породам (хлор-апатит, нередко скаполиты, в псевдоморфозах по основным плагиоклазам, а в сублиматах при вулканических извержениях - хлориды железа, свинца и других металлов).
При эндогенных процессах минералообразования фтор и хлор в конце концов связываются исключительно с петрогенными элементами. Изредка легкорастворимые Хлориды Fe, Мn и Сu встречаются как неустойчивые продукты возгона в районах вулканической деятельности.
Среди фторидов петрогенных элементов широко распространен как самостоятельный минерал флюорит - наиболее труднорастворимый из фтористых соединений. Однако хлоридов петрогенных элементов эндогенного происхождения мы не знаем (опять-таки за исключением продуктов возгона при вулканических извержениях: NaCl, КСl и др.)
Несмотря на то что в гидротермальных месторождениях мы не встречаем самостоятельных минералов хлора, все же имеются определенные указания на то, что эти растворы были обогащены растворимыми соединениями хлора. Об этом свидетельствует состав устанавливаемых в кристаллах жидких включений маточного раствора, захваченного еще в процессе роста кристаллов. В некоторых из них даже наблюдались микроскопические кристаллы NaCl и других хлоридов, указывающие на высокую концентрацию хлористых соединений в этих жидких включениях, а следовательно, и в гидротермальных растворах. Нормально выкристаллизовавшиеся хлориды в гидротермальных образованиях отсутствуют, очевидно, потому, что высокая концентрация ионов в растворе, необходимая для выпадения этих легкорастворимых хлоридов в виде кристаллов, не могла быть достигнута. Причина этого, по всей вероятности, заключается в том, что в концентрациях соленого раствора, выполнявшего трещинные полости, и растворов, заполнявших поры вмещающих пород, существовала большая разница. Это различие в концентрациях и могло обусловить сильную диффузию ионов, а вместе с тем и снижение концентрации хлоридов в остаточных гидротермальных растворах.
Изучение парагенетических соотношений минералов в некоторых оловорудных и железорудных месторождениях приводит к выводу о том, что галоиды (фтор и хлор) при пневматолито-гидротермальных процессах могли играть серьезную роль в переносе ряда металлов (в виде летучих соединений: SnF4, SiF4, FeCl3 и др.). Эти металлы, обладая высоким сродством с кислородом, при реакциях с Н2O в условиях более низких давлений и температур или при увеличении концентрации кислорода в растворах, очевидно, способны менять свои анионы F1- и Сl1- на O2- с образованием весьма устойчивых окислов: SnO2, SiO2, Fe2O3, Fe3O4 и др. (экспериментальным путем эти реакции уже давно были изучены). Если освобождающиеся при этом ионы фтора встречают ионы кальция, то тут же отлагается флюорит. В противном случае фтор уходит из сферы действия реакции. В случае разложения хлористых соединений ионы хлора, естественно, уже не могут задержаться в сколько-нибудь значительном количестве в виде минералов, поскольку все соединения его с петрогенными элементами представляют легкорастворимые вещества. Тем не менее во многих контактово-метасоматических и гидротермальных месторождениях железа, особенно в случае нахождения скаполитов и других хлорсодержащих минералов, участие хлора в привносе металлов теперь представляется гораздо более вероятным, чем это допускали раньше.
Бор, углерод и фосфор располагаются в левой части рассматриваемой группы элементов (рис. 370).
О легкой подвижности соединений бора в остаточных эндогенных растворах можно судить прежде всего по нахождению минералов этого элемента, в частности турмалина, в пегматитах и гидротермальных образованиях и, значительно реже, датолита, аксинита, людвигита и других в контактово-метасоматических, иногда гидротермальных месторождениях. В продуктах вулканических эксгаляций установлены фториды щелочей и бора (ферручит - NaBF4 и авогадрит - KBF4), а горячие водяные пары нередко в растворенном состоянии содержат борную кислоту.
Насколько существенную роль играет бор в переносе тяжелых металлов при процессах рудообразования, сказать трудно. Изредка встречаются в значительных скоплениях бораты Fe и Мn и крайне редко двойной борат кальция и олова-норденшельдит (CaSnB2O6). Однако в лабораторных условиях установлено, что боро-медные и боро-молибденовые комплексные соединения обладают необычайной летучестью.
Экзогенные водные полибораты, главным образом Na и Са, в виде значительных скоплений распространены в бороносных соляных залежах, чаще в районах вулканической деятельности.
Углерод в эндогенных образованиях широко распространен в виде карбонатов главным образом петрогенных элементов, а также Fe и отчасти Мn, редких земель, Ва и др. (кальцит, доломит, анкерит, сидерит, родохрозит, паризит, витерит и др.). Все они возникают при гидротермальных процессах минералообразования и часто сопровождают рудные минералы в жилах, выделяясь нередко в последнюю очередь. В виде комплексного аниона [СO3]2- углерод входит в состав некоторых силикатов, образующихся в контактово-метасоматических месторождениях и пегматитах (скаполит, канкринит). Изредка среди пегматитов встречаются мелкие скопления графита, иногда углеродистых веществ в соединении с U, Th и TR (тухолит, карбуран, карбоцер). Имеются указания на жидкие органические соединения в минералах гидротермальных жил. В карбонатах и кварце описаны включения жидкой углекислоты в виде капелек, захваченных, очевидно, во время роста кристаллов. Наконец, обильные выделения газообразной СO2 иногда вместе с СО (окисью углерода) и другими газообразными продуктами, происходят в сольфатарах и, особенно, мофеттах в районах действующих вулканов. Все это говорит о том, что углерод принимает участие в более поздних процессах минералообразования.
При экзогенных процессах происходят мощные накопления углерода в виде торфов, бурых и каменных углей, нефтей и карбонатных осадочных толщ. Источником этих накоплений служат продукты широко развивающихся на земной поверхности биохимических реакций в растительном мире, поглощающем огромные массы углекислоты из атмосферы в процессе фотосинтеза.
Фосфор в таблице химических элементов (рис. 370) стоит рядом с кремнием. Хотя в природе и встречаются фосфаты, содержащие в виде изоморфной примеси анион [SiO4], а также силикаты, равным образом содержащие анион [РO4], все же фосфор в геохимическом отношении существенно отличается от кремния. Несмотря на то что кларк фосфора в 200 с лишним раз меньше кларка кремния, в природе устанавливается большое количество разнообразных фосфатов. Среди них главное значение имеет фосфат кальция - апатит и его разновидности (с ним связано около 95% всего находящегося в земной коре фосфора). В виде мельчайших акцессорных включений апатит почти всегда наблюдается в изверженных горных породах, причем относительно более богаты основные и щелочные породы. В генетической связи со щелочными породами известны крупнейшие скопления апатито-нефелиновых руд.
В больших количествах (по сравнению с горными породами) фосфаты распространены в пегматитах; характерно, что в ассоциации с турмалином и другими борсодержащими минералами они, как правило, не встречаются. В типичных гидротермальных месторождениях фосфаты редки.
При экзогенных процессах в коре выветривания и, особенно, в осадочных известковистых породах образуются иногда мощные скопления, главным образом в виде фосфоритов. Роль фосфора в растительном и животном мире общеизвестна.
Элементы группы железа. Согласно таблице (рис. 370) эта группа обнимает элементы: Sc, Ti V, Cr, Mn, Fe, Ni и Co, - характеризующиеся особым строением атомов (переменное число электронов, свойственное второй с периферии оболочке).
Элементы этой группы обладают той особенностью, что по своему поведению в земной коре занимают как бы промежуточное положение между петрогенными и металлогенными элементами. С одной стороны, все эти элементы в виде изоморфных примесей входят в состав силикатов основных и ультраосновных изверженных пород. Общее количество их в таком рассеянном состоянии достигает огромных цифр. С другой стороны, мы знаем многочисленные месторождения кислородных соединений этих металлов, например, хромистого железняка - в ультраосновных породах с высоким содержанием Cr и Fe, титаномагнетитовых руд (Fe, Ti, V) - в основных породах. Известны и месторождения богатых железом сульфидных руд в основных породах с промышленным содержанием Ni и Со (в пентландите). Лишь марганец при эндогенных процессах очень редко образует промышленные скопления, и то преимущественно в виде жил гидротермального происхождения. Что касается скандия, то этот элемент в рассеянном состоянии присутствует преимущественно в железо-магнезиальных силикатах и особенно в железных рудах, хотя единственный в природе скандиевый минерал - тортвейтит (Sc2Si2O7) был встречен в гранитных пегматитах Норвегии.
Титан, железо и марганец также нринимают участие в составе пегматитовых образований, связанных с богатыми щелочами изверженными породами. Они входят в состав многочисленных редких минералов, образуя сложные окислы с Nb, Та, Zr, редкими землями и др. Для железа известны также крупные контактово-метасоматические месторождения магнетитовых руд.
Кроме того, железо чрезвычайно широко представлено в виде сульфидов в обычных гидротермальных месторождениях (пирит, пирротин, арсенопирит и др.). Гораздо реже встречаются сульфиды и арсениды Ni и Со. Крайне редки в виде сульфидов V и Мn. Для элементов правой части группы весьма характерно образование дисульфидов (MnS2, FeS2, NiS2), диарсенидов (FeAs2, NiAs2, CoAs2), сульфоарсенидов и сульфоантимонидов (FeAsS, NiSbS и др.). Распространены, наконец, также карбонатные руды Fe и Мn (сидерит, родо-хрозит), нередко в виде жильных месторожений.
В экзогенных условиях осадочным путем из коллоидных растворов в морских бассейнах образуются крупные месторождения железа и марганца. В. И. Вернадский в образовании этих руд придает большое значение особым микроорганизмам. В коре глубокого выветривания ультраосновных (магнезиальных) пород возникают гидросиликатные руды никеля и отчасти кобальта. Скопления ванадия в виде ванадатов (ванадинит, деклуазит и др.) встречаются в зонах окисления некоторых свинцовоцинковых сульфидных месторождений.
Группа редких металлов. Здесь объединяются следующие элементы: Y, La и редкие земли (№ 58-71), имеющие много общего между собой в геохимическом отношении, затем Zr, Hf, Nb и Та. Все эти элементы в тех или иных соотношениях встречаются совместно в редких минералах, представляющих собой сложные по составу окислы, образующиеся преимущественно в богатых щелочами изверженных породах-нефелиновых сиенитах. Таковы минералы группы перовскита (кнопит, дизаналит, лопарит), минералы группы пирохлора, а также такие минералы, как фергюсонит, самарскит, эшинит, эвксенит, эвдиалит и др. Характерной особенностью состава этих минералов является обилие изоморфных примесей не только элементов, входящих в эту группу, но и ряда элементов из окружающих ее в таблице групп. Часто в значительных количествах присутствуют такие элементы, как Na, Са, Sr, в меньшей степени - Мn, Fe, Sc, а также Th, U, Sn, Ti, Sb, W и др. Если мы обратимся к рис.142 (стр.201), то увидим, что все элементы но размерам ионных радиусов легко разбиваются на две определенные группы и участвуют в сложных составах этих минералов в соответствии с условиями гетеровалентыого изоморфизма.
Кроме того, в гранитных и сиенитовых пегматитах распространены и более простые по составу минералы элементов рассматриваемой группы: циркон, иногда содержащий Th и U; монацит - фосфат редких земель, нередко содержащий Th; ксенотим - фосфат иттрия; колумбит, танталит, реже карбонаты редких земель (группа паризита), фториды редких земель (группа флюорита), ортит, апатит, содержащий редкие земли в виде изоморфной примеси к Са, и др.
Поведение элементов этой группы и минералов, их содержащих, при экзогенных процессах изучено, к сожалению, еще очень недостаточно.
Группа радиоактивных элементов. Эта группа располагается в нижней части геохимической таблицы (рис. 370). Главное значение из них имеют Ra, Th и U. Четырехвалентные Th и U, обладающие большими размерами ионных радиусов, при эндогенных процессах способны образовать труднорастворимые ортосиликаты (торит, ураноторит) и входят в виде изоморфной примеси в состав циркона, а также сложных окислов, упомянутых выше. Кроме того, они, особенно U4+, распространены в виде окислов (торианита и уранинита). Характерно, что в случае нахождения уранинита в пегматитах в него, в виде изоморфной примеси, часто входит Th, чего не наблюдается в типичных гидротермальных месторождениях уранинита.
Уран, как известно, в окислительных условиях способен давать шестивалентные ионы. Соединения, образованные ионами такого типа, сравнительно легко растворимы в воде и богато представлены в виде минералов экзогенного образования (фосфаты, арсенаты, вападаты, силикаты, карбонаты, сульфаты).
Что касается Ra, то мы не знаем для него самостоятельных минералов. Как продукт радиоактивного распада он присутствует в определенном соотношении к урану во всех урановых минералах. Характерной геохимической особенностью этого элемента является то, что он, как крупный двухвалентный катион, способен давать, так же как и барий, труднорастворимый сульфат. Поэтому не удивительно, что он нередко входит в виде изоморфной примеси в состав барита. В зонах окисления богатых сульфидами урановых месторождений отношение Ra : U несравненно выше, чем в первичных рудах.
Группа молибдена и вольфрама (рис. 370) примыкает с правой стороны к группе редких металлов и снизу к группе элементов железа: Mo, W, Тс и Re. Несмотря на то что в таблице Менделеева два первых элемента находятся в одной и той же группе и характеризуются почти одинаковыми размерами атомов и ионов, они имеют совершенно разные геохимические особенности. Молибден в эндогенных природных условиях в виде катионов Мо4+ обладает столь сильным сродством с серой, что наблюдается почти исключительно в виде сульфида (MoS2), и притом в парагенетической связи с самыми различными минералами (сульфидами и окислами): пирротином, пиритом, халькопиритом, кварцем (чаще всего), кальцитом, шеелитом, вольфрамитом, магнетитом (без сопровождающих других сульфидов) и силикатами трехвалентного железа (эпидотом и др.). В редких случаях, очевидно в особо окислительной обстановке, он входит как катион Мо6+ в виде изоморфной примеси к вольфраму в шеелит (CaWO4).
Вольфрам же, наоборот, в эндогенных условиях проявляет сильно выраженное сродство с кислородом, встречаясь почти исключительно в виде вольфраматов (вольфрамита и шеелита). Даже в сплошных сульфидных рудах этот элемент представлен кислородными соединениями (вольфрамитом).
Оба металла (молибден и вольфрам) генетически связаны с кислыми изверженными породами и образуют промышленные месторождения примерно в одних и тех же условиях либо в виде кварцевых жил вблизи апикальных частей интрузивов, либо в виде контактово-метасоматических залежей в скарнах. Однако выпадение их из растворов в виде минералов происходит в силу разных причин: вольфрамит и шеелит, очевидно, начинают выпадать из восходящих растворов, как только в них будет достигнута соответствующая концентрация кислорода, а молибденит - при необходимой концентрации ионов S2-, возникающих в процессе диссоциации H2S в гидротермальных растворах. Вероятно, этим объясняются нередко устанавливаемые возрастные соотношения и пространственная разобщенность минералов Мо и W, даже в тех случаях, когда они наблюдаются в одних и тех же рудных телах (например, в скарнах).
В экзогенных условиях молибденит разлагается с переходом Мо4+ в Мо6+ и образованием молибдатов Са (новеллит) и Рb (вульфенит в зонах окисления свинцово-цинковых месторождений). Заметные концентрации Мо иногда устанавливаются в некоторых осадочных породах, глинистых сланцах, содержащих органические вещества. Вольфрамит более устойчив, но при окислении входящих в его состав двухвалентных Мn и Fe также разлагается с образованием гидроокислов этих металлов и тунгстита (HWO4).
Рений самостоятельных минералов не образует. В виде изоморфной примеси, как выяснилось, он чаще всего входит в состав молибденита. При процессах выветривания ион Re4+ окисляется до Re7+, что и служит причиной разобщения молибдена и рения в экзогенных образованиях.
Элементы группы платины. Эти элементы (Ru, Rh, Pd, Os, Ir и Pt), тесно связанные друг с другом по условиям нахождения, в природе встречаются в виде самородных металлов. Как уже указывалось при описании минералов группы платины, Pt и Pd, располагающиеся в правой части триад, в природных условиях ведут себя резко отлично от Ru и Os, занимающих места в левой колонке. Элементы Rh и Ir играют двойственную роль: с одной стороны, они входят в состав твердых растворов с Pt и Pd, нередко вместе с Fe, Ni и Сu, а с другой - образуют химические соединения переменного состава с Os и Ru, кристаллизующиеся в гексагональной сингонии. Несмотря на это, парагенетически те и другие минеральные виды тесно связаны между сообой.
Следует упомянуть, что палладий отличается от других элементов группы платины тем, что в природных условиях способен давать твердые растворы и интерметаллические соединения с типичными металлогенными элементами, расположенными в таблице правее группы платины. Таковы, например, палладистое золото (порпецит), станнопалладинит (Pd3Sn2), стибиопалладинит (Pd3Sb). Для платины известно соединение PtTe3 (нигглиит). Наконец, установлены крайне редкие сульфиды и арсениды: PtS (куперит), (Pt, Pd, Ni) S (брэггит), RuS2 (лаурит), PtAs2 (сперрbлит). Эти минералы парагенетически связаны уже с типичными сульфидными рудами, главным образом с халь-копиритом, пирротином, пентландитом, миллеритом и др.
Группа типичных металлогенных элементов сульфидных месторождений. Это так называемые купро-ионы, характеризующиеся 18-электронной оболочкой, начиная от Сu и кончая Рb (рис. 370). Все они обладают сильной поляризующей способностью. Характернейшей геохимической их особенностью является резко выраженная склонность к образованию сульфидов (за исключением Аu), свидетельствующая о сильном сродстве их с серой.
В левой части этой группы располагаются медь, серебро и золото, примыкающие к группе элементов платины и железа. Нужно заметить, что они обладают и некоторыми общими свойствами с ними. Все они в природе встречаются в самородном виде. Медь с Fe, Ni и Со часто образует двойные сульфиды (халькопирит, борнит, минералы группы линнеита).
Для меди и серебра бросается в глаза та особенность, что они наряду со свинцом образуют главную массу сульфосолей: сульфоарсенитов, сульфоан-тимонитов и сульфовисмутитов (прустит, пираргирит, блеклые руды, энаргит, айкинит и др.)
В ряду Сu, Ag, Аu степень сродства с серой падает от Сu и Аu. Этим объясняется тот факт, что в халькозине под микроскопом устанавливались мельчайшие включения самородного серебра, что определенно указывает на большее сродство меди с серой. Золото в природных условиях вообще не известно в виде сульфидов, хотя парагенетически тесно связано с сульфидами других металлов. Необходимо, между прочим, упомянуть, что серебро очень часто присутствует в галените (нередко до десятых долей процента) в виде включений главным образом сульфосолей и теллуридов. Главная масса серебра в настоящее время получается попутно со свинцом.
Из двухвалентных катионов наибольшее значение по распространенности имеют цинк и свинец, простые сульфиды которых (ZnS и PbS) поразительно тесно связаны друг с другом в гидротермальных месторождениях.
Однако это не означает того, что оба эти элемента одинаковы в геохимическом отношении. Об этом можно судить хотя бы по тому факту, что свинец, в отличие от цинка, образует многочисленные и разнообразные сульфосоли (буланжерит, джемсонит и др.) иногда в виде двойных соединений с катионами Сu1+ и Ag1+. Оба элемента в эндогенных условиях встречаются также в виде силикатов, но силикаты Рb гораздо более разнообразны, хотя наблюдаются несравненно реже силикатов Zn. Затем, окислы цинка - цинкит (ZnO), ганит (Zn,Al2O4), франклинит и др. - встречаются исключительно в эндогенных образованиях, а окислы Рb - массикот (РbО), глет (РbО) и сурик (Рb3O4)- в ассоциации с самородным свинцом, правда крайне редко, наблюдались в зонах окисления некоторых свинцовых месторождений, и т. д.
Кадмий, индий и галлий не образуют самостоятельных минералов, за исключением кадмия, который в крайне редких случаях встречается в виде сульфида-гринокита (CdS) и карбоната-отавита (CdCO3) в экзогенных образованиях. Обычно кадмий рассеян в виде изоморфной примеси в светлых, маложелезистых сфалеритах (до десятых долей, изредка до единиц процента). Кроме того, кадмий связан и с минералами кальция, в частности с флюоритом (размеры ионов Са и Cd почти одинаковы).
Индий чаще всего с помощью спектрального анализа устанавливается в черных, богатых железом сфалеритах, так называемых сульфостаннатах (в цилиндрите 0,1-1,0%) и сульфогерманатах. В повышенных количествах иногда встречается в халькопирите. При экзогенных процессах рассеивается. Изредка накапливается в таких минералах, как каламин, смитсонит и ярозит.
Галлий рассеян главным образом в сфалеритах, где его содержание обычно достигает тысячных, реже сотых и изредка десятых долей процента. В виде Ga2O3 он известен в минералах алюминия: в мусковите, затем гидраргиллите, бёмите, диаспоре (главных составных частей бокситов). В одном случае в германите (Cu3GeS4) было установлено содержание галлия 1,85%.
Германий в природных условиях образует, правда очень редко, самостоятельные минералы: германит (Cu3Ge S4), аргиродит (Ag8GeS6), а также накапливается в некоторых сульфостаннатах: канфильдите (Ag8 Sn S6), франкеите (Pb5Sn3Sb2S14). В ничтожных количествах присутствует в виде примеси в некоторых силикатах, сложных окислах (самарските, гадолините, танталите), сульфидах, чаще в сфалерите, вюртците (иногда до 0,3%) и минералах олова, в золах каменных углей (до 0,01%, реже до 0,1%).
Олово по своему поведению в природных условиях несколько отличается от других элементов рассматриваемой группы. В восстановительных условиях среди сульфидных руд оно образует сернистые соединения двухвалентного- герценбергит (SnS), тиллит (PbSnS2), станнин, франкеит (Pb5Sn3Sb2S14) - и четырехвалентного олова-канфильдит (Ag8SnS6), цилиндрит (Pb3Sn4Sb2S14). Однако в более обогащенной кислородом обстановке, в том же сульфидном типе месторождений, олово, в силу ярко выраженного сродства с кислородом, кристаллизуется уже в виде окисла - касситерита в ассоциации с теми же сульфидами (пирротином, пиритом, сфалеритом, халенитом, станнином, халькопиритом, арсенопиритом и др.).
Весьма характерно, что при воздействии остаточных, обогащенных кислородом растворов на ранее отложившиеся сульфиды олова происходит их разложение с образованием в виде псевдоморфоз тонких смесей, например, галенита с касситеритом на месте тиллита (Pb Sn S2), или халькопирита с касситеритом на месте станнина и др. В некоторых месторождениях имеют место л обратные случаи (замещение станнином ранее выделившеюся касситерита), называющие на восстановительный характер реакции в последующие моменты минералообразования.
В кварцево-касситеритовом типе месторождений олово распространена обычно в виде крупных кристаллов касситерита в парагенезисе с кварцем, флюоритом, иногда топазом, турмалином, вольфрамитом и другими минералами. Сульфиды или не встречаются, или имеют подчиненное значение. Пара-генетические соотношения минералов заставляют предполагать, что касситерит в этом типе месторождений мог образоваться в результате разложения иного типа летучих соединений, по всей вероятности галоидных.
В гранитных пегматитах касситерит нередко содержит в себе в виде примесей Nb, Та, Zr, Мn, Fe, W, иногда в виде мельчайших включений соответствующих минералов, напоминающих по расположению продукты распада твердых растворов (ср. размеры ионов на рис. 142). О вхождении олова в виде примеси в сложные окислы Nb, Та, Zr, Ti и др. уже было упомянуто ранее. Следует добавить, что для олова известны также силикаты и бораты в виде двойных солей. С помощью спектрального анализа олово в ничтожных количествах устанавливается во многих минералах, преимущественно в сульфидах и силикатах.
С геохимической точки зрения, представляют особый интерес нахождение самородного олова в россыпях, приуроченных к ультраосновным изверженным породам, а также интерметаллические соединения Sn с Pd и Pt (станнопалладикит и другие, еще слабо изученные минералы), генетически связанные вместе с сульфидами с основными изверженными породами.
Ртуть по своим свойствам существенно отличается от располагающихся в той же группе периодической системы элементов Zn и Cd. Нестойкость химических соединений ртути и способность легко переходить в самородное состояние сближает этот элемент с золотом. Не случайны поэтому встречающиеся в природе амальгамы золота, а также серебра и палладия. С другой стороны, некоторые химические свойства (способность давать селениды, теллуриды, оксигалоидные соединения и др.) сближают этот элемент с Рb (в соединениях двухвалентного катиона). В отличие от него, ртуть редко образует сульфосоли: ртутьсодержащую блеклую руду и ливингстонит (HgSb4S7).
Главнейшим минералом ртути в земной коре является, как известно, киноварь (HgS), образующаяся вследствие высокой упругости ее паров в условиях низких температур и давлений, нередко в ассоциации с антимонитом, иногда реальгаром, флюоритом и др. В ничтожных количествах в рассеянном состоянии ртуть присутствует во многих сульфидах, а также в таких минералах, как барит (повидимому, в одновалентном состоянии), флюорит и др.
Таллий в геохимическом отношении изучен очень плохо. Несмотря на очень низкий кларк, в земной коре он наблюдается в виде очень редких самостоятельных минералов: крукезита - (Cu,Tl,Ag)2Se, лорандита - TlAsS2, врбаита - Tl(As,Sb)3S5 и утчинсонита -(Cu,Ag,Tl)2S•PbS•2As2S3? Последние три минерала встречаются в парагенезисе с реальгаром, аурипигментом, антимонитом, марказитом, пиритом и др. Характерно, что пирит и, особенно, марказит некоторых низкотемпературных гидротермальных месторождений содержат до нескольких десятых процента таллия.
В экзогенных условиях сульфиды металлов рассматриваемое группы чрезвычайно легко разлагаются (за исключением киновари) и образуют различные кислородные соли: сульфаты (Рb, Сu), карбонаты (Сu, Zn, Рb), фосфаты, арсенаты, ванадаты (Рb, Сu, Zn), силикаты (Zn, Сu), а также окислы (Sn, Сu, Рb, Hg) и изредка галогениды (Ag, Сu, Hg).
Группа полуметаллов и тяжелых минерализаторов: As, Sb, Bi, Se и Те. Полуметаллы - мышьяк, сурьма и висмут - обладают рядом характерных свойств. Прежде всего, эти элементы в виде комплексных сульфоанионов с катионами предыдущей группы, главным образом c Cu1+,Agl+ и Рb2+, образуют большое число сульфосолей: сульфоарсениты, сульфоантимониты и сульфовисмутиты. Затем следует отметить сульфиды этих элементов: AsS, As2S3, Sb2S3 и Bi2S3.
Мышьяк и в меньшей степени сурьма образуют ряд простых соединений с элементами VIII группы таблицы Менделеева (особенно триады Fe, Со, Ni): сульфоарсениды (FeAsS, CoAsS и др.), сульфоантимониды (NiSbS), диарсениды (FeAs2, PtAs2, CoAs2), арсениды (NiAs) и редкие антимониды (NiSb). Висмут уже не дает подобных соединений.
Наконец, полуметаллы обнаруживают склонность легко давать в восстановительных условиях самородные металлы, причем эта склонность резко увеличивается с возрастанием атомного веса и общей поляризации ионов (от As к Bi). Кроме того, в природе известны интерметаллические соединения с Сu (Cu6Sb, Cu3As и др.), Ag (Ag3As, Ag3Sb), Аu (Au2Bi) и примыкающими к ним слева Ni (Ni3As) и Pd (Pd3Sb).
Для висмута следует отметить еще одну замечательную особенность, а именно его способность в ряде соединений изоморфно замещать Рb (см. таблицу - рис. 370), указывающую на близость химических свойств этих элементов. Известны месторождения висмутоносного галенита* с октаэдрической отдельностью вдоль тончайших пластинок висмутина, очевидно как продукта распада твердого раствора. В некоторых висмутовых месторождениях в самом висмутине (однородном под микроскопом) содержатся существенные количества свинца. Известны, наконец, такие интерметаллические соединения как Ni3(Bi,Pb)2S2, (паркерит), в котором Bi изоморфно замещается Рb (экспериментально проверен изоморфный ряд Ni3Pb2S2- Ni3Bi2S2).
*(В месторождении Серро-де-Паско (Перу) висмут как побочный продукт при плавке свинцовых концентратов добывается в столь значительных количествах, что это месторождение занимает первое место в мире по добыче висмута. )
В экзогенных условиях рассматриваемые полуметаллы ведут себя весьма различно. В то время как для сурьмы в продуктах выветривания сульфидных месторождений характерны окислы, для мышьяка мы преимущественно наблюдаем арсенаты Fe••, Fe•••, Ni, Со, Сu, Zn, Pb, Mg, Са и др. Висмут обычно дает карбонаты, изредка другие типы соединений.
Что касается селена и теллура, то в поведении этих двух элементов в природных условиях хотя и имеются черты сходства, но гораздо более резко выражены черты различия. Селен практически является аналогом серы и в виде изоморфной примеси в незначительных количествах часто входит в состав многих сульфидов (галенита, пирита, висмутина и др.). Самостоятельные селениды редки (известны для Сu, Ag и Hg). Теллур преимущественно наблюдается в виде самостоятельных минералов, главным образом теллуридов Bi, Pb, Hg, Au, Ag, Сu, изредка Ni и Pt (см. таблицу - рис. 370). Особенно характерна связь теллура с благородными металлами, с которыми он в сущности образует интерметаллические соединения (называемые теллуридами). Кроме того, теллур встречается в самородном состоянии. Эти геохимические особенности селена и теллура вполне соответствуют их положению в периодической системе элементов Менделеева.
В зоне окисления в редких случаях устанавливаются селениты (Сu, Ni, Со), теллуриты (Fe•••), селенаты (Рb) и теллураты (Fe, Hg, Bi). Селен и здесь большей частью в виде изоморфной примеси входит в состав сульфатов.
Группа тяжелых галоидов (Вr и J) располагается в самом конце геохимической таблицы. Эти элементы в виде редких бромидов и иодидов встречаются в зонах окисления некоторых рудных месторождений, и притом с такими сильно поляризующими металлами, как Ag, Сu, Hg. В особо окислительной обстановке в условиях пустынного климата образуются растворимые иодаты (Сu, Са, Рb). Главные массы Вr и J находятся в рассеянном состоянии, повидимому в качестве изоморфной примеси к хлорсодержащим минералам.
При экзогенных процессах бром накапливается в остаточных рассолах соляных озер, сильно обогащенных магнием и содержащих ионы К и Cs. Иод в существенных количествах устанавливается в некоторых продуктах жизнедеятельности растений, а также нефтяных водах и грязевых вулканах.
|
ПОИСК:
|
При использовании материалов проекта обязательна установка активной ссылки:
http://geoman.ru/ 'Физическая география'