Глава III. О методах детальных минералогических исследований
В студенческой практике, для того чтобы научиться простыми методами определять распространенные в земной коре минералы, приходится иметь дело с более или менее крупными кристаллами или же с однородными минеральными массами. Для этой цели обычно пользуются внешними диагностическими признаками изучаемых минералов, описанными в главе II. Многие из минералов, особенно те, которые могут содержать ценные для промышленности металлы, требуют дополнительных исследований с помощью паяльной трубки и элементарных качественно-химических реакций, излагаемых в специально составленных определителях.
Однако более редко встречающиеся или трудно определимые простыми методами минералы, обнаруживаемые при систематических минералогических исследованиях, могут быть достоверно определены лишь при условии применения более совершенных методик. Необходимость этих детальных исследований появляется во всех случаях, когда возникает задача более полного познания состава природных образований:
- при петрографическом изучении горных пород, необходимом для составления геологических карт;
- при освоении какого-либо нового месторождения, в связи с чем ставится задача всестороннего изучения вещественного состава руд с целью решения вопросов о комплексном использовании всех компонентов сырья;
- при специальных исследованиях в районах, особо интересных в минералогическом отношении;
- при решении вопросов геохимии, требующих углубленных исследований минерального вещества, и т. д.
При детальном изучении минералов в случае необходимости применяют обычно следующие методы исследования: кристаллохимический, рентгенометрический , кристаллооптический, термический и химикоаналитический совместно с спектральным и иногда рентгеноспектральным анализом. Все эти методы подробно излагаются в специальных руководствах. Мы здесь укажем лишь, к чему они сводятся и когда применяются.
Кристаллохимический анализ, разработанный Е. С. Федоровым, может быть применен в тех случаях, когда мы имеем дело с кристаллами, и притом достаточно крупными, чтобы можно было измерить углы между гранями кристаллов на гониометре*. Этим путем удается не только установить сингонию и вид симметрии кристаллов, но и определить состав минерала. Е. С. Федоровым создан монументальный труд "Царство кристаллов" (1920), содержащий специальные таблицы по кристаллохимическому анализу.
*(Для мелких кристалликов А. Н. Заварицким был предложен способ гониометрических исследований с помощью "федоровского столика" (Зап. мин. общ., 1929, стр. 280-297))
В последнее время его школой разрабатываются определители кристаллов с учетом углов между гранями, комбинаций простых форм и оптических констант. У нас в Союзе начал выходить в свет многотомный "Определитель кристаллов", составляемый коллективом авторов при Ленинградском горном институте.
Рентгенометрический анализ, применяемый для определения вещества путем сравнения рентгенограмм, может быть произведен различными методами, из которых наиболее употребительны: метод вращения кристалла(Поляни), метод рентгеновского гониометра (Вейссенберга) и метод порошка (Дебая).
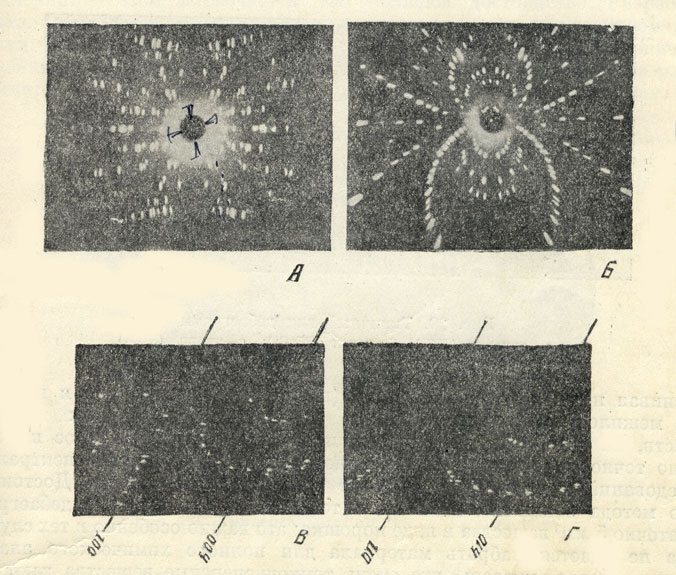
Рис. 28. Вверху слева (А) приведена рентгенограмма, полученная при облучении вращающегося кристалла монохроматическими рентгеновскими лучами; справа (Б) - лауэграмма, полученная при облучении неподвижного кристалла непрерывным спектром рентгеновской трубки. Внизу приведены рентгенограммы полученные по методу Вейссенберга (для хромовой слюды - фуксита): В-разрешение нулевой слоевой линии (h 0l) и Г-разрешение первой слоевой линии (h 11)
Первые два метода применяются в тех случаях, когда мы имеем дело с монокристаллами. Под методом вращения кристалла подразумевается рентгеносъемка вращающегося кристалла при облучении монохроматическими рентгеновскими лучами. В противоположность этому, в методе Лауэ кристалл неподвижен и облучается непрерывным спектром рентгеновской трубки. Примеры рентгенограмм приведены на рис. 28. При более широко применяемой рентгеносъемке по методу Вейссенберга, помимо вращения кристалла, производится также поступательное движение цилиндрической пленки параллельно оси вращения кристалла, т. е. перпендикулярно к рент геновскому лучу. Вид рентгенограмм, полученных по методу Вейссенберга, весьма характерен по расположению интерференционных пятен вдоль кривых (рис. 28); они позволяют сравнительно легко (геометрическим путем) производить расчет индексов.

Рис. 29. Типичный вид дебаеграмм а-сильвин (KCl), б-галит (NaCl), в-кварц (SrO2)
Метод Дебая обладает тем важным преимуществом, что позволяет изучать агрегатные массы минерала, включая скрытокристаллические и тонкодисперсные вещества, и поэтому широко применяется в практике минералогов с целью диагностики. Рентгенограмма, называемая обычно дебаеграммой (рис. 29), получается в специальной камере на полоске светочувствительной пленки, на которой после проявления видны различной интенсивности линии-дужки (части колец, создаваемых конусами рентгеновских лучей, отраженных от наиболее плотно упакованных плоскостей в кристаллических осколках растертого испытуемого вещества).
Сравнивая полученную дебаеграмму (по интенсивности линий и вычисленным межплоскостным расстояниям) с дебаеграммами других известных веществ, на которые похоже по внешним признакам испытуемое вещество, можно точно определить данный минерал, имея результаты спектральных исследований и хотя бы некоторые оптические константы. Достоинство этого метода заключается также в том, что для получения дебаеграммы достаточно 5 мм3 вещества в виде порошка; это важно особенно в тех случаях, когда не удается набрать материала для полного химического анализа. Следует, однако, указать, что очень тонкодисперсные вещества дают слабо выраженные, завуалированные дебаеграммы, а аморфные тела (в собственном смысле) вообще не обнаруживают отражения рентгеновских лучей.
В связи с этим упомянем также об электронографическом методе исследования тончайших пленок толщиной в несколько миллимикронов или чрезвычайно тонкодисперсных коллоидных масс. Этот метод основан на свойстве электронов закономерно рассеиваться при встрече с закономерно расположенными атомами, т. е. аналогично тому, что мы имеем для рентгеновских лучей. Разница заключается лишь в том, что рентгеновские лучи проходят в глубь кристаллического вещества, тогда как электронный пучок способен проникать в глубину всего лишь до 0,01μ (т. е. до одной стотысячной миллиметра). Получаемые при этом методе электронограммы имеют тот же вид, что и дебаеграммы.
Кристаллооптический анализ сводится к определению с помощью микроскопа ряда оптических констант, свойственных изучаемому минералу.
Прозрачные минералы горных пород и руд исследуются в тонких шлифах (толщиной около 0,02 мм) или в виде порошков. К числу оптических констант, подлежащих определению в изучаемом минерале, относятся: показатель преломления N (для оптически изотропных минералов) или главные показатели преломления Ng, Nm и Np (для анизотропных минералов), устанавливаемые с помощью специально подобранных иммерсионных жидкостей или более точно на микрорефрактометре; затем двупреломление Ng-Np, оптический знак (для анизотропных минералов), угол оптических осей 2V (для анизотропных двуосных минералов) и др. Этот метод определения прозрачных минералов с помощью поляризационного микроскопа в проходящем свете в последнее время достиг высокой степени совершенства, особенно для плагиоклазов, разработка методики определения которых на универсальном столике Е. С. Федорова (рис. 30) была завершена А. Н. Заварицким. Этим методом могут быть точно определены даже мельчайшие кристаллические зерна (диаметром в несколько сотых миллиметра), устанавливаемые в тонких прозрачных шлифах в виде включений. В этом заключается его большое достоинство. Разработаны специальные определители прозрачных минералов под микроскопом в виде таблиц. Они широко используются петрографами при микроскопическом изучении горных пород. Применение этого метода требует овладения работой на микроскопе и усвоения ряда специальных приемов исследования.

Рис. 30. Универсальный столик Е. С. Федорова
Непрозрачные минералы, слагающие главным образом руды месторождений металлических полезных ископаемых, а также встречающиеся в виде включений в горных породах, изучаются в зеркально отполированных шлифах в отраженном свете под микроскопом с помощью специального осветителя, называемого опак-иллюминатором. К числу оптических констант относятся: показатель отражения R (способность минерала отражать то или иное количество падающего света, измеряемая с помощью микрофотометрокуляра или фотоэлемента), а для оптически анизотропных двуосных минералов - главные показатели отражения Rg, Rm и Rp, двуотражение Rg-Rp и др. Методика определения оптических констант для анизотропных, особенно двуосных, минералов еще не разработана. Тем не менее определение показателя отражения в комбинации с данными определения прочих свойств изучаемых под микроскопом минералов (твердость, цвет, отношение к реактивам и др.) оказывает большую услугу при изучении руд под микроскопом в отраженном свете. Этим путем во многих случаях могут быть определены даже мельчайшие включения рудных минералов размером в тысячные доли миллиметра.
Термический анализ, введенный в практику исследований акад. Н. С. Курнаковым, сводится к получению кривых нагревания (или охлаждения) вещества с целью установления эндо- и экзотермических эффектов, обусловливаемых физическими и химическими превращениями, происходящими в исследуемом веществе при повышении температуры (выделение воды, окисление, восстановление, переход в новую полиморфную модификацию и др.).
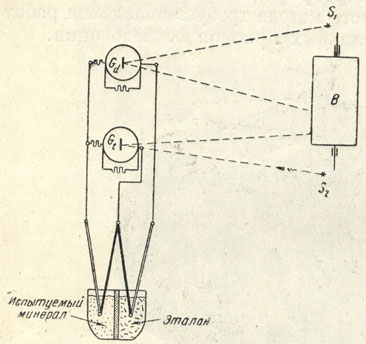
Рис. 31. Схема диференциальной и температурной записи термограмм с помощью самопишущего пирометра Н. С. Курнакова S1 и S2- источники света; В-барабан; Gd и Gt-гальванометры диференциальной и простой термопар. Сплошной жирной линией показана платинородиевая проволока, двойной линией - платиновая проволока и тонкой линией-медная проволока
Запись кривых нагревания производится обычно автоматически при помощи регистрирующего пирометра, соединенного с комбинированной (простой и диференциальной) термопарой, опущенной в тигель, разделенный специальной перегородкой на две части (рис. 31). В одну часть помещается измельченный испытуемый минерал, в другую - какое-либо эталонное индиферентное вещество (MgO, Al2O3 и др.). В ту и другую цепь включены зеркальные гальванометры (Gt и Gd), каждый из которых посылает свою световую точку на светочувствительную бумагу, натянутую на медленно вращающийся при помощи часового механизма барабан В*.
*(Запись производится в темной комнате; результаты записи проявляются как обычные фотоотпечатки )
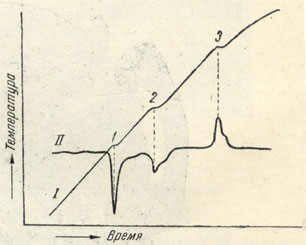
Рис. 32. Термограммы I-вались обыкновенной термопарой; II-запись диференциальной термопарой
В процессе нагревания гальванометр простой термопары на светочувствительной бумаге записывает ход температурной кривой (рис. 32, кривая I), на которой в случае происходящих превращений в нагреваемом веществе фиксируются местные изменения наклона (1) или горизонтальные площадки (2), отвечающие эндотермическим эффектам (с поглощением тепла), или скачки вверх (3), свидетельствующие об экзотермических эффектах (с выделением тепла), происходящих при нагревании испытуемого вещества. Гальванометр, включенный в цепь диференциальной термопары, регистрирует только разность двух термоэлектродвижущих сил, т. е. разность температур индиферентного эталона и исследуемого вещества в процессе одновременного их нагревания. Поэтому на кривой диференциальной записи (рис. 32, кривая II) получаются более резко выраженные пики с вершинами, направленными вниз (эндотермические эффекты) и вверх (экзотермические эффекты), явно указывающие на превращения, происходящие в испытуемом веществе. В этом и заключается смысл получения диференциальной записи процесса нагревания (или охлаждения).
В минералогической практике этот метод обычно применяется при исследовании трудно определимых на глаз или другими способами скрытокристаллических и тонкодисперсных масс. Для ряда минеральных образований (каолина, гидратов глинозема, гидроокислов железа, карбонатов, хлоритов и др.) получаются характерные кривые нагревания, способствующие определению минеральных видов.
Необходимо отметить, что само число минералов, для которых этим методом удается получить какие-либо характерные данные, имеющие диагностическое значение, составляет относительно небольшой процент от числа известных в природе минералов. К ним преимущественно относятся химические соединения, содержащие воду, гидроксил и углекислоту. Затем этим методом удается узнать природу лишь основной массы исследуемого вещества. Механические примеси, которые нас в большинстве случаев интересуют в испытуемых минеральных массах, при содержании их до 5-10%, за некоторыми исключениями, не устанавливаются.
С другой стороны, в ряде случаев при изучении минеральных веществ возникает необходимость более полного познания их свойств, особенно когда эти вещества приобретают практическое значение. Бывает важно точно знать, что происходит с данным веществом при нагревании. Для этой цели получение только кривых нагревания является недостаточным. Продукты, получающиеся в результате каждого установленного превращения вещества, требуют химического анализа, изучения оптических свойств и рентгенометрических исследований.
Важно знать точно температуры, при которых происходят эти превращения. Последнему требованию термограммы, как выяснилось, не всегда удовлетворяют: регистрация этих превращений самопишущими приборами обычно запаздывает, причем разница достигает 60-100° и больше. В этом отношении для минералов, содержащих воду и гидроксил, гораздо более точные данные можно получить из кривых дегидратации (обезвоживания) минералов при нагревании. Для этой цели испытуемое вещество в количестве 1-2 г или более, предварительно взвешенное вместе с платиновым тиглем, выдерживается в электрической печи последовательно при определенных температурах (с интервалом обычно 50°) до тех пор, пока потеря веса по сравнению с предыдущим взвешиванием не станет меньше 0,03-0,05%, и только после этого температура печи повышается на следующую ступень. Полученные таким путем кривые потери воды дают ясное представление о том, при каких температурах наступают превращения в веществе.
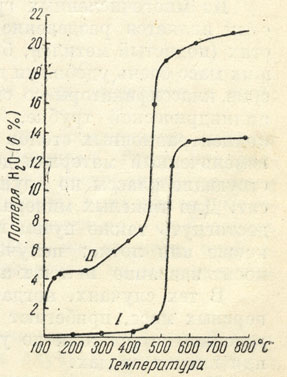
Рис. 33. Кривые обезвоживания каолинита (I) и галлуазита (II)
На рис. 33 приведены две кривые обезвоживания каолинита - Al4[Si4O10] [OH]8 и галлуазита - Al4[Si4O10] [OH]8•4H2O. В то время как для каолинита (кривая I), не содержащего воды, а только гидроксильные группы ОН, сильные изменения происходят в интервале температур 500-550°, в галлуазите (кривая II) молекулы кристаллизационной воды выделяются до 150° (первый скачок кривой), а гидроксильные группы - при температуре 450-500° (второй, высокий скачок кривой вверх). Как устанавливается рентгенометрическими исследованиями, с потерей гидроксильных ионов кристаллические решетки этих минералов разрушаются, показатель преломления сильно падает.
Химический анализ является сравнительно трудоемким и дорогим методом исследования. Поэтому к полным химическим анализам прибегают в тех случаях, когда имеются основания установить какую-либо новую разновидность или новый минерал, по ряду свойств отличающийся от известных минералов, либо когда без данных химического анализа невозможно решить вопрос о разновидности изучаемого минерала, обладающего переменным составом, либо в тех случаях, когда минерал принадлежит к числу редких соединений, для которых известно ограниченное число полных анализов, и т. д.
Количество чистого, т. е. освобожденного от примесей, вещества, необходимое для полного химического анализа, должно составлять, как минимум, 1-2 г. что не всегда удается набрать, особенно для редко встречающихся и рассеянных в породе или руде мелких кристалликов или зерен минерала.
Если исследуемый минерал наблюдается в виде мелких кристаллических друз в пустотках, то его обычно предварительно отбирают каким-либо способом, например с помощью стальной иглы, всаженной в деревянную ручку. Полученную таким путем массу тщательно сортируют под бинокулярной лупой с помощью той же иглы, отбирая интересующий нас минерал в необходимом количестве для химического анализа и других видов исследований. Если минерал наблюдается вкрапленным в породу в значительном количестве, то породу в целом подвергают дроблению и измельчению, отсеивая каждый раз мелочь через сито с отверстиями 0,5, 1,0 мм или крупнее (в зависимости от размеров зерен вкрапленного минерала). Отборка минерала тем же способом производится под бинокулярной лупой.
В случае если минерал является акцессорным, т. е. крайне редко вкрапленным в породу, приходится прибегать к получению концентратов тем или иным механическим способом. При этом используется либо разница в удельных весах минералов (гравитационные методы), в магнитных свойствах (методы магнитной сепарации), либо отношение минералов к флотационным реагентам (методы флотации), к электричеству (электростатические методы) и т. д.
Из многочисленных гравитационных методов обогащения самым простым является разделение зерен минералов в тяжелых или вязких жидкостях (йодистый метилен, бромоформ, жидкость Туле и др.). В случае больших масс очень удобными для этой цели являются лабораторные гидравлические классификаторы со спирально восходящей струей воды в стеклянной цилиндрической трубке, а также лабораторные небольшие столы типа концентрационных столов Вильфлея или др. Для работы на этих приборах измельченный материал должен быть предварительно разбит на соответствующие классы по крупности зерна с помощью специально подобранных сит. Для тяжелых минералов той же цели, но в более грубом виде, можно достигнуть также путем промывки дробленого материала в старательском ковше или лотке; получающаяся при этом фракция тяжелых минералов носит название шлиха.
В тех случаях, когда приходится изучать состав землистых тонкодисперсных масс, прибегают к отмучиванию в стеклянных банках или к разделению на фракции по удельным весам с помощью центрифуги в жидких или вязких средах.
Из методов магнитной сепарации в лабораторной практике наибольшим распространением пользуется разделение минералов в тяжелых фракциях с помощью электромагнитов. Регулируя силу электротока, питающего катушки, и расстояния между полюсами, удается получать из шлихов речных россыпей до 10-15 почти мономинеральных фракций, очистить которые обычными путями не представляет большого труда.
Подготовленный для химического анализа материал предварительно должен подвергнуться спектральному анализу, если он не был произведен ранее. Этот анализ необходим для того, чтобы знать, какие химические элементы вообще содержатся в минерале и какие из них могут быть определены при химическом анализе. Следует заметить, что эти определения с помощью спектрографа производятся быстро, и притом в некоторых количественных соотношениях для элементов. Это и важно знать, прежде чем начать химический анализ.
Данные полного химического анализа, выраженные в весовых процент тах, необходимо пересчитать на атомные (молекулярные) количества, с тем чтобы можно было вывести химическую формулу минерала. С этой целью данные весовых количеств каждого элемента (окисла) делят на его атомный вес ("молекулярный вес" окисла)*. Полученные числа должны показывать, в каком отношении находятся данные элементы (или окислы), входящие в состав минерала. Необходимо указать, что соотношения компонентов, вычисляемые по данным химических анализов, никогда не бывают строго кратными в силу или недостаточно высокой точности самих анализов, или других причин. Приведем для иллюстрации два примера.
*(Атомные веса берутся по таблице Менделеева. Молекулярный вес окисла со ставляется из суммы атомных весов элементов; например, для SiO2 он равен 28,1 + 2 х 16,0 = 60,1 )
| % по весу | Атомный вес | Атомное количество | Отношение | |
|---|---|---|---|---|
| Pb | 42,75 | 207,2 | 0,204 | 1 |
| Cu | 12,77 | 63,6 | 0,201 | 1 |
| Sb | 24,76 | 121,8 | 0,206 | 1 |
| S | 19,40 | 32,0 | 0,606 | 3 |
| Сумма 99,68 | ||||
Таким образом, химическая формула минерала должна выразиться в виде: PbCuSbS3.
| % по весу | Атомный вес | Атомное количество | Отношение | ||
|---|---|---|---|---|---|
| SiO2 | 46,06 | 60,1 | 0,767 | 1 | |
| Al2O3 | 0,11 | 101,9 | 0,001 | - | |
| Fe2O3 | нет | - | - | - | |
| FeO | 1,83 | 71,8 | 0,025 | 0,772 | 1 |
| MnO | 44,76 | 70,9 | 0,630 | ||
| CaO | 6,59 | 56,1 | 0,117 | ||
| Сумма 99,35 | |||||
Химическая формула этого минерала может быть выражена в виде: (Мn,Са)О•SiO2 или (Mn,Ca)SiO3.
Бывают случаи, когда не удается отобрать для химического анализа совершенно свободный от посторонних примесей минерал. Тогда при расчете данных химического анализа, если количество посторонней примеси невелико и минералогическая природа ее установлена, приходится вычислять состав интересующего нас минерала приблизительно, сообразуясь с микроскопическими данными исследований. Если же примеси присутствуют в больших количествах, то в этом случае пересчеты на минеральный состав не всегда будут внушать доверие.
Спектральный анализ в практике минералогических исследований стал применяться настолько широко, что во многих случаях заменяет метод паяльной трубки, особенно в лабораторных условиях. Этот метод определения присутствующих в минерале химических элементов основан, как известно, на том, что каждый химический элемент при достаточном нагревании испускает лучи определенных, характерных для него длин волн, устанавливаемые с помощью спектроскопа. Главными преимуществами спектрального анализа являются точность и быстрота определения содержащихся в минерале катионов металлов. Особенно это имеет значение при определении таких ценных редких металлов, как олово, молибден, индий, германий, галлий, кадмий и др. Мало того, для ряда металлов разработана методика определения примерного количественного значения содержания. Количество материала, требующегося для анализа, может быть ограничено несколькими миллиграммами. В Этом также заключается важное достоинство этого метода.
Рентгеноспектральный (рентгенохимический) анализ основан на том, что испытуемое вещество, помещенное на поверхность антикатода, при действии катодных лучей испускает рентгеновы лучи определенной длины волны для каждого из содержащихся в нем химических элементов, т. е. аналогично-тому, что мы имеем при обычном спектральном анализе. Важно лишь, чтобы напряжение, приложенное к электродам рентгенотрубки, было достаточным для возбуждения видимых лучей, характерных для того или иного элемента. Рентгеноспектральный анализ особенно ценен при количественном определении в минералах редких земель: Y, Nb, Та, Hf, Re, определение которых обычными химическими методами составляет чрезвычайно трудоемкую задачу.
Наконец, необходимо указать на важность экспериментальных химических и физико-химических исследований с целью получения в лабораторных условиях искусственных соединений, отвечающих по составу природным соединениям-минералам. Этим путем, по крайней мере в некоторых случаях, удается получить совершенно аналогичные но внешним формам и составу соединения (например, урановые слюдки) и выяснить таким путем условия образования и кристаллизации природных соединений. В этом отношении для минералогов должны представлять исключительный интерес также исследовательские и экспериментальные работы химических и физико-химических институтов, занимающихся проблемами использования минерального сырья.
|
ПОИСК:
|
При использовании материалов проекта обязательна установка активной ссылки:
http://geoman.ru/ 'Физическая география'